„Die äußeren Segmente der Stäbchenzellen in noch nie dagewesenem Detailreichtum abbilden“ – Ein Interview mit Matthias Pöge vom Max-Planck-Institut für Biochemie
Grundlagenforschung zur Retina: Ein Team des Max-Planck-Instituts für Biochemie (MPIB) hat in Zusammenarbeit mit der University of California, Irvine (UCI) erstmals auf molekularer Ebene Strukturen der Membranarchitektur des äußeren Segments der Stäbchenzellen visualisiert – mit Hilfe der Kryo-Elektronentomographie. Ein Interview mit Matthias Pöge, Postdoc am MPIB, dem Erstautor der Studie.

Die Membranarchitektur des äußeren Stäbchensegments, des spezialisierten sensorischen Ziliums der Stäbchen-Photorezeptorzellen, bildet die Grundlage für den ersten Schritt des Sehens – die Phototransduktion. Die Kryo-Elektronentomographie ermöglicht jetzt neue, detaillierte Einblicke in die Stäbchenzellen und damit in die Mechanismen, die den Pathologien bestimmter Genmutationen zugrunde liegen, die zur Erblindung führen.
Das Max-Planck-Institut für Biochemie in Martinsried bei München zählt zu den führenden internationalen Forschungseinrichtungen auf den Gebieten der Biochemie, Zell- und Strukturbiologie sowie der biomedizinischen Forschung. Hier erforschen rund 350 Wissenschaftlerinnen und Wissenschaftler die Struktur von Proteinen – an einzelnen Molekülen, aber auch an komplexen Organismen. Ein weiterer wichtiger Forschungsaspekt ist die biomedizinische Grundlagenforschung.
Inwiefern hat die Kryo-Elektronentomographie die technische Grundlage für Ihre Forschungsergebnisse geliefert?
Matthias Pöge: Tatsächlich hätten wir unsere Untersuchungen mit keiner anderen Methode durchführen können, um zu diesen Ergebnissen zu kommen. Wir waren an der molekularen Architektur der äußeren Stäbchenzellensegmente interessiert. Dabei handelt es sich um ein hochgeordnetes Membransystem. Es besteht aus hunderten fast identisch geformten, abgeflachten Membranvesikeln, die im immer etwa gleichen Abstand weniger Nanometer übereinandergestapelt sind. Diese Membrandisks selbst und ihre zytosolischen Zwischenräume sind vor allem mit den Proteinen der Phototransduktions-Maschinerie gefüllt, also jenen Proteinen, die ein Lichtsignal in ein Nervensignal umwandeln.
Die klassischen bzw. länger etablierten Methoden der Strukturbiologie wie die Röntgenkristallographie oder NMR-Spektroskopie versuchen in der Regel, die einzelnen Komponenten aufzureinigen und isoliert zu untersuchen. Das ist jedoch oftmals nur bedingt aussagekräftig. Wir wollten uns das äußere Stäbchenzellensegment in seiner vollen Komplexität so naturnah und detailliert wie möglich anschauen. Dafür ist die Kryo-Elektronentomographie, meines Erachtens, die momentan stärkste und am besten geeignete bildgebende Methode.
Könnten Sie an dieser Stelle auf das Prinzip der Kryo-Elektronentomographie eingehen?
Sehr gerne. Die Kryo-Elektronentomographie erlaubt es uns, eine Vielzahl der Proteine innerhalb unserer Zellen in ihrer natürlichen Umgebung dreidimensional zu visualisieren. Sie öffnet uns gewissermaßen ein Fenster in das Zellinnere. Bei der Kryo-Elektronentomographie handelt es sich um eine technisch sehr komplexe Methode. Daher würde ich zum besseren Verständnis gern etwas weiter ausholen. Der Name setzt sich aus drei Komponenten zusammen: Kryo, Elektronen und Tomographie. Auch wenn das etwas kryptisch klingen mag, ist der Name für die Methode wirklich zutreffend.
Dann lassen Sie uns doch mit den Elektronen anfangen.
Klar. Bei unseren Forschungen schauen wir uns das Innere der Zelle im Elektronenmikroskop an. Genauer gesagt im Transmissionselektronenmikroskop, denn diese Methode verspricht viel mehr Details aufzulösen als das mit einem Lichtmikroskop möglich wäre. Das klingt nun erst einmal gut, ist aber mit einem hohen technischen Aufwand verbunden.
Im Elektronenmikroskop benötigen wir ein Ultrahochvakuum, um Bilder zu generieren. Unsere Zellen bestehen aber zum Großteil aus Wasser. Wenn wir diese nun dem Vakuum im Elektronenmikroskop aussetzen, würde das Wasser schlagartig verdampfen und die Zelle schlicht platzen. Um das zu verhindern, werden die Zellen im klassischen Verfahren chemisch fixiert, gefärbt und in ein Kunstharz eingebettet. Danach werden von dieser fixierten Probe dünne Schnitte angefertigt, die im Elektronenmikroskop stabil sind. Seit den frühen 40er Jahren können Wissenschaftler:innen mit dieser klassischen Präparationsmethode tief in das Innere der Zelle blicken, was unser heutiges Verständnis über die zelluläre Ultrastruktur maßgeblich geprägt hat.
Allerdings führt dieser Präparationsprozess vor allem auf molekularer Ebene zu Artefakten. Zum Beispiel werden Membranen deutlich deformiert. Zudem denaturieren Proteine im Harz, sie aggregieren zum Teil oder werden deplatziert. Wir interessierten uns aber genau dafür – die Anordnung und die Wechselwirkungen der Proteine im natürlichen Zustand der Zelle. Mit der klassischen Methode können wir das nicht untersuchen.
Und Kryo bezieht sich auf die spezifische Art der Probenpräparation?
Genau. Da das Problem im Hochvakuum des Elektronenmikroskops das Wasser im flüssigen Aggregatzustand ist, könnte man die Probe auch ganz einfach einfrieren. Allerdings dehnt sich das Wasser beim Einfrieren durch die Bildung von Eiskristallen aus, was die Zellen wiederum zerstört. Die Lösung ist extrem schnelles Einfrieren. Deshalb wurden unsere Proben im Bruchteil einer Sekunde auf fast -200 °C herunter gekühlt. So bleibt dem Wasser buchstäblich keine Zeit, um Eiskristalle zu formen. Bei dieser Methode erstarrt die Probe in einem glasähnlichen Zustand, man spricht auch von Vitrifizierung. Das erhält die Zelle auf molekularer Ebene im naturähnlichsten Zustand, der technisch momentan möglich ist.
In der Regel sind vitrifizierte Zellen noch zu dick, um direkt brauchbare Bilder im Elektronenmikroskop aufzunehmen. Mithilfe eines fokussierten Ionenstrahls schneiden wir deshalb 100-300 Nanometer dünne Scheibchen aus der Probe. Diese so genannten Lamellen laden wir dann zur Datenaufnahme ins Elektronenmikroskop.
Dann bleibt noch die Tomographie.
So ist es. Die Bilder, die wir im Transmissionselektronenmikroskop generieren, sind, in guter Näherung, 2D Projektionen unsere Probe. Diese können jedoch trügerisch sein. Wie bei einem Schattenspiel, wenn wir die Finger unserer Hand in bestimmter Weise Anordnen und aus dem richtigen Winkel an eine Wand projizieren, erscheint das Bild wie ein Hund oder ein Schmetterling. Drehen wir unsere Hand jedoch etwas zur Seite, wird die Illusion zerstört. Schattenspiele sind ein schönes Beispiel für Projektionsbilder, die uns in die Irre führen. Blicken wir über Projektionsbilder aus dem Elektronenmikroskop in das Innere der Zelle, müssen wir solche Fehlinterpretationen jedoch unbedingt vermeiden. Dazu kippen wir unsere Probe im Elektronenmikroskop und nehmen diese Projektionsbilder aus verschiedenen Projektionswinkeln auf (Abbildung 1 A). Mit dieser Bilderserie (Abbildung 1 B) können wir dann eine 3D Repräsentation unserer Probe erstellen, was man ein Tomogramm nennt (Abbildung 1 C). Diese analysieren wir, je nach Fragestellung, mit unterschiedlicher Software am Computer.
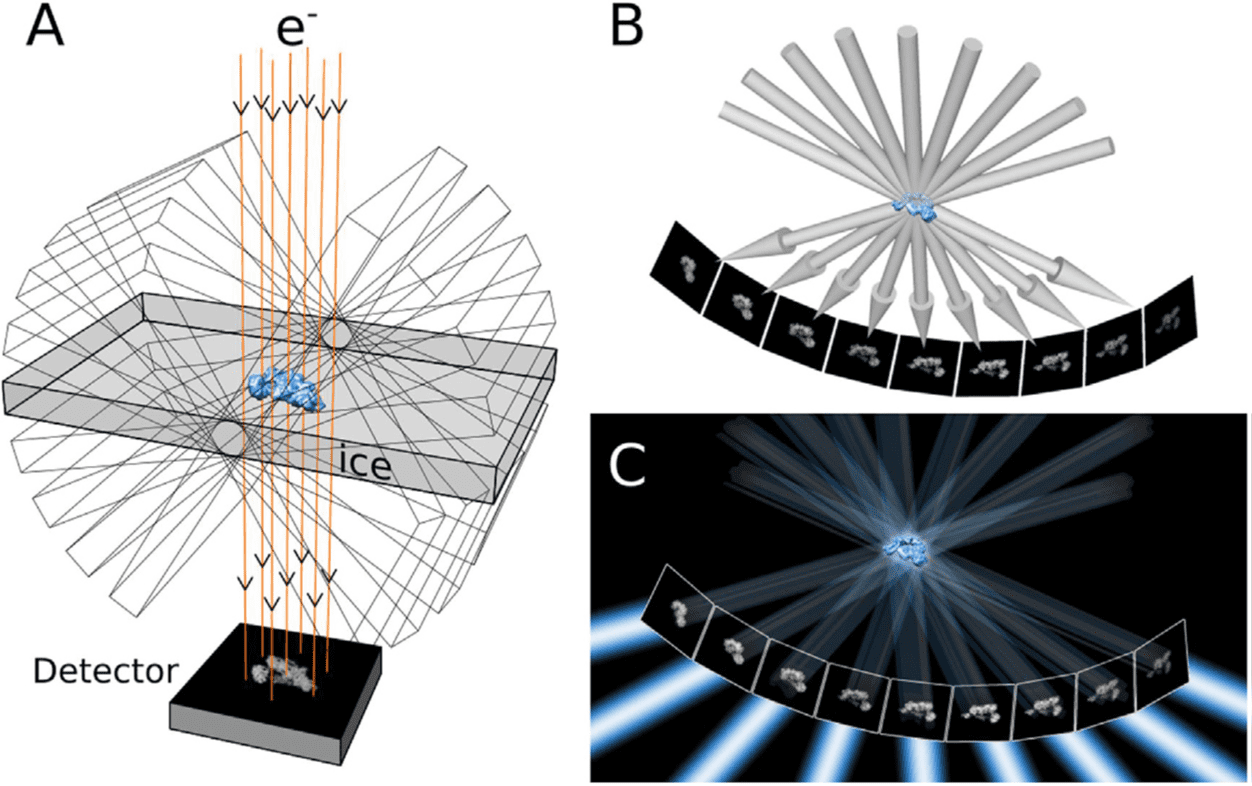
Abbildung 1: Das Prinzip der Elektronentomographie: Die Probe wird im Transmissionselektronenmikroskop relativ zum Elektronenstrahl (e-) gekippt und mehrere Projektionsbilder von unterschiedlichen Probenorientierungen aufgenommen (A). Diese Bilderserie (B) wird genutzt, um durch Rückprojektion eine 3D Repräsentation der Probe zu rekonstruieren – ein Tomogramm (C). Die Abbildung wurde aus Advances in cryo-electron tomography for biology and medicine, Koning et al., Annals of Anatomy, 2018 übernommen. Ice = vitrifiziertes Wasser.
Welche Strukturen konnten Sie mit Hilfe der Kryo-Elektronentomographie in der Netzhaut entdecken?
Tatsächlich konnten wir die äußeren Stäbchenzellsegmente in noch nie dagewesenem Detailreichtum abbilden (Abbildung 2). In unseren Tomogrammen sehen wir eine Vielzahl von Proteinen im Membranstapel, insbesondere zwischen den einzelnen Membrandisks. Hierbei handelt es sich zum Großteil um Proteine der Phototransduktions-Maschinerie. Momentan ist es jedoch noch nicht möglich, diese Proteinstrukturen tatsächlich Transduzin, Phosphodiesterase 6 oder anderen Proteinen des äußeren Stäbchenzellensegments zuzuordnen.

Abbildung 2: Schnitt eines Kryo-Elektronentomogramms des äußeren Stäbchenzellensegments. Der Schnitt ist etwa 1 Nanometer dick und enthüllt die hochgeordnete Membranarchitektur des äußeren Segments sowie zahlreiche Proteine zwischen den Membrandisks. DL = Disklumen; DM = Diskmembran; Zy = Zytosol; PM = Plasmamembran; DR = Diskrand.
Unsere wichtigste Entdeckung ist ein Proteingerüst am Rand der Membrandisks, welches höchst wahrscheinlich an der Organisation der dortigen Membrankrümmung beteiligt ist (Abbildung 3). Die Entstehung dieser außergewöhnlich hohen Membrankrümmung beschäftigt Wissenschaftler:innen seit der ersten strukturellen Beschreibung des äußeren Stäbchenzellensegments kurz nach dem zweiten Weltkrieg.
Seit den 90er Jahren verdichten sich die Hinweise, dass Proteinkomplexe aus Peripherin-2 und ROM1 dafür verantwortlich sind. Unter anderem hat man diese Komplexe aufgereinigt und in Membranvesikeln rekonstituiert. Das führte zur Bildung von Membranzylindern, die durch ein Proteingerüst organisiert wurden. Wir konnten uns jetzt direkt anschauen, wie dieses Gerüst in der Zelle aufgebaut ist. In unseren Tomogrammen haben wir tausende kleinere Subtomogramme entlang der Diskränder ausgeschnitten, diese dann aufeinander ausgerichtet und gemittelt. So konnten wir das Proteingerüst am Diskrand, welches in den Tomogrammen nur erahnbar ist, mit einer Auflösung von unter 2 Nanometern darstellen.
Zusätzlich haben wir die 3D Struktur eines Peripherin-2 Dimers von der DeepMind Software AlphaFold2 vorhersagen lassen, welche hervorragend in die Untereinheiten unseres Proteingerüsts passt. Das ist noch nicht der ultimative Beweis, dass das Gerüst tatsächlich aus Peripherin-2 und ROM1 Komplexen besteht. Aber unsere Resultate und die Ergebnisse, die viele andere Forscher:innen in Jahrzehnte langer Arbeit erbracht haben, fügen sich jetzt sehr gut zusammen. Wie ein Puzzleteil, dass endlich am richtigen Platz sitzt.
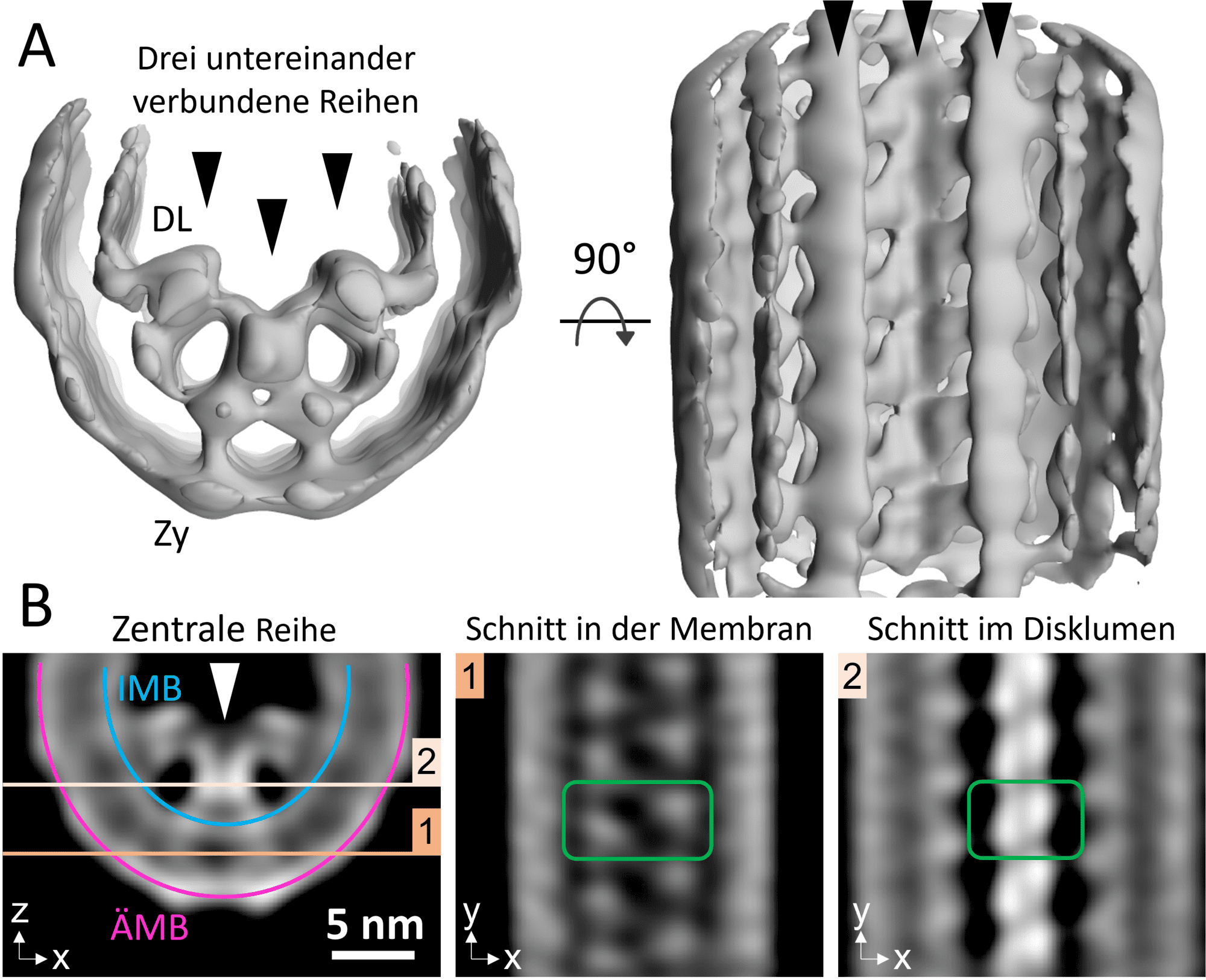
Abbildung 3: Die Mittelung tausender Subtomogramme des Diskrandes enthüllt ein Proteingerüst bestehend aus drei fortlaufenden und untereinander verbundenen Reihen von Untereinheiten. (A) Isoflächenrepräsentation (Fläche gleichen Grauwertes) in der Frontalansicht entlang des Diskrandes (Links in A) und als Draufsicht vom Disklumen auf den Diskrand (Rechts in A). DL = Disklumen; Zy = Zytosol. Schwarze Pfeilspitzen zeigen die drei Reihen an. (B) Orthogonale Schnitte des Proteingerüsts. Jeder Schnitt hat eine Dicke von weniger als 0,3 Nanometern. Links in (B) ist ein xz-Schnitt abgebildet. Die blaue Linie markiert das innere Membranblättchen (IMB), die pinke Linie das äußere Membranblättchen (ÄMB) und die weiße Pfeilspitze die zentrale Reihe. Die beiden orangenen Linien deuten die Positionen der xy-Schnitte durch die Membran (Mitte in B) und durch das Disklumen (Rechts in B) an. Die grünen Rahmen markieren das Signal einer Untereinheit entlang der zentralen Reihe. Jede Untereinheit besitzt zwei separate Transmembrandichten (Mitte in B). Im Disklumen, hingegen, stehen die Untereinheiten in engem Kontakt.
In der Zelle besteht das Gerüst aus nur drei fortlaufenden und untereinander vernetzten Reihen dieser Untereinheiten und erstreckt sich entlang des gesamten Diskrandes (Abbildung 3 A). Dieser Aufbau unterscheidet sich wesentlich von dem, was die früheren in vitro Experimente vermuten ließen. Außerdem trägt das Gerüst in der zellulären Umgebung zu einer deutlich höheren Membrankrümmung bei. Das ist meiner Meinung nach auch die Stärke und das Faszinierende and der Kryo-Elektronentomographie. Sie erlaubt es uns direkt zu sehen, wie sich gewisse Membran- oder Proteinstrukturen in der Zelle genau organisieren. Im Reagenzglas verhalten sich dieselben aber isolierten Komponenten oftmals ganz anders.
Welche Rolle könnte diese Entdeckung für die Ophthalmologie im Allgemeinen, aber auch für Entwicklung neuer Therapien für Erkrankungen wie Retinitis pigmentosa und Morbus Stargardt spielen?
In unserer Abteilung betreiben wir hauptsächlich Grundlagenforschung. Wir waren in erster Linie an der molekularen Architektur des gesunden Auges bzw. des äußeren Stäbchenzellensegments interessiert. Daraus können wir aber Rückschlüsse ziehen, warum gewissen Mutationen, also im Krankheitsfall, zu Problemen in unserem Körper führen. Zum Beispiel gibt es eine Vielzahl von Peripherin-2 Mutationen, die zur Erblindung führen. Heute wissen wir, dass die meisten dieser Mutationen in der extrazellularen Domäne dieses Proteins liegen. Unsere Datenanalyse deutet darauf, dass das Proteingerüst am Diskrand hauptsächlich durch die extrazelluläre bzw. diskluminale Domäne seiner Untereinheiten zusammengehalten wird (Abbildung 3 B). Somit erscheint es uns naheliegend, dass diese Peripherin-2 Mutationen das Zusammenfügen des Proteingerüsts stören, damit die strukturelle Integrität des äußeren Segments beeinträchtigen und somit zur Erkrankung führen. Ich bin davon überzeugt, dass dieses Grundlagenwissen Mediziner:innen in Zukunft helfen wird Krankheiten besser zu verstehen und dadurch auch neue Therapien zu entwickeln.
Studie: Determinants shaping the nanoscale architecture of the mouse rod outer segment (eLife)