„Schießscheibenmakulopathie sowie eine Verdünnung sämtlicher Netzhautschichten“ - Dr. Frank Stehr über die Kinderdemenz NCL
Augenärzte, Orthoptisten und Optometristen spielen bei der Diagnose der auch als Kinderdemenz bezeichneten Neuronalen Ceroid Lipofuszinose (NCL) eine entscheidende Rolle. Dr. Frank Stehr, Vorstand der NCL-Stiftung, über die Symptomatik und Diagnose dieser neurodegenerativen Erkrankung sowie das Engagement der Stiftung für die Betroffenen und ihre Familien.

Um was für Erkrankungen handelt es sich bei den Neuronalen Ceroid Lipofuszinosen (NCL)?
Dr. Frank Stehr: Die Kinderdemenz NCL umfasst eine Gruppe genetisch bedingter neurodegenerativer Erkrankungen, die meist im ersten Lebensjahrzehnt auftreten. Die Kinder erblinden, entwickeln eine Epilepsie und es kommt durch das Absterben der Nervenzellen im Gehirn zu einem fortschreitenden Abbau der geistigen und motorischen Fähigkeiten. NCL ist bisher nicht heilbar und führt zu einem frühen Tod, meist noch vor dem 30. Lebensjahr. NCL gilt als seltene Erkrankung, in Deutschland gibt es ca. 700 Betroffene, weltweit ca. 70.000.
Mit welchen dieser Subtypen werden Augenärzte, Orthoptisten und Optometristen am häufigsten konfrontiert?
Hier ist vor allem die juvenile NCL zu nennen, nach genetischer Nomenklatur auch als CLN3 bezeichnet. Das Vorstellungsalter der betroffenen Kinder liegt bei 5 - 8 Jahre. Im typischen Fall treten bei einem zuvor gesunden Kind um die Zeit des Schulanfangs Sehschwierigkeiten auf und es kommt zu einem rasch voranschreitenden beidseitigen Visusverlust. Den oben genannten Fachgruppen kommt daher bei der NCL-Diagnose eine entscheidende Rolle zu.
Bei welchen Symptomen sollte man an die Möglichkeit einer NCL-Erkrankung denken?
An NCL sollte gedacht werden, wenn bei Visusverlust, Sprachentwicklungsverzögerung, Stillstand oder Rückschritte der psychomotorischen Entwicklung und Epilepsie mindestens zwei dieser Symptome kombiniert auftreten.
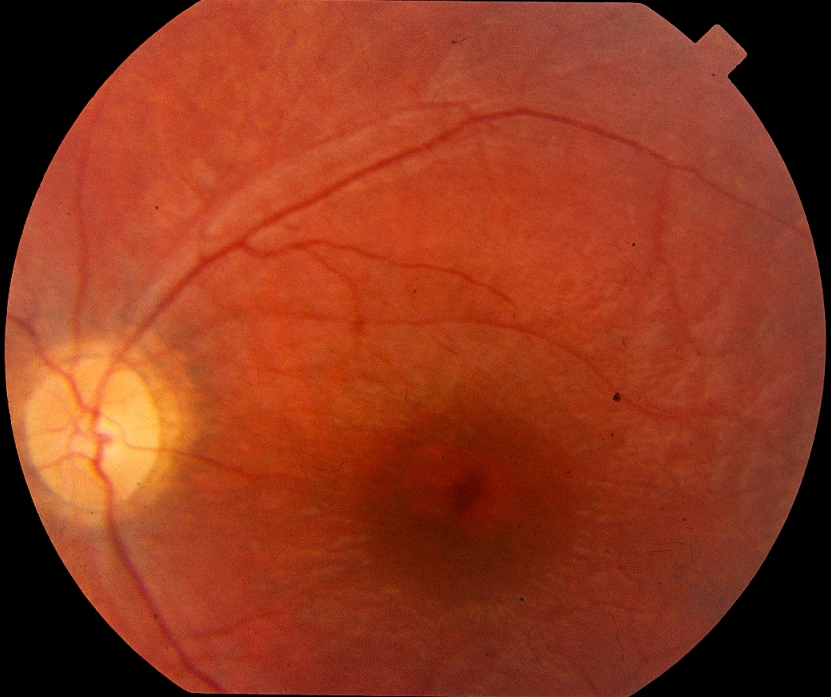
Augenhintergrund eines NCL-Patienten mit typischer Schießscheibenmakulopathie (so genanntes „Bull’s eye“). © NCL-Stiftung
Wie erfolgt dann die Diagnose?
Bei einem Verdachtsfall wird die Diagnose letztendlich mittels molekulargenetischer Analyse in einem humangenetischen Labor gestellt.
Woraus resultiert der Visusverlust bei NCL?
Hierbei handelt es sich primär um eine Degeneration der Photorezeptoren und sekundär auch der Bipolarzellen, die die Information von den Photorezeptoren zu den Ganglienzellen übermitteln. Früh tritt dabei auch eine Degeneration der Makula auf, die das Sehen von zentralen Objekten beeinträchtigt.
Was fällt bei der augenärztlichen/optometrischen Untersuchung auf?
Zu nennen ist im Augenhintergrund eine Schießscheibenmakulopathie sowie bei der OCT der Netzhaut eine Verdünnung sämtlicher Netzhautschichten mit weitgehendem Verlust der Photorezeptorschicht zentral und peripher. Ebenfalls ist bereits kurze Zeit nach dem Beginn der Erkrankung eine erhebliche konzentrische Gesichtsfeldeinschränkung zu erkennen. Zur Bestätigung der retinalen Degeneration ist ein Ganzfeld-Elektroretinogramm sinnvoll. Auch, dass bei NCL früh eine Degeneration der Makula auftritt, die das Sehen von zentralen Objekten beeinträchtigt – also ein Zentralskotom – kann spezifisch mittels multifokalem ERG verifiziert werden. Auffällig ist dabei bei den Kindern das typische Phänomen des Vorbeischauens beim Fixieren von Objekten, das so genannte „Overlooking“. Beide ERG-Potenziale sind bei fortgeschrittener Progression nicht mehr nachweisbar.
Welche Möglichkeiten der Therapie bestehen?
Die Entwicklung kausaler Therapieansätze ist herausfordernd, da NCL eine heterogene Gruppe an erblichen Erkrankungen darstellen und bisher noch nicht bei allen NCL-Formen der eigentliche Pathomechanismus aufgeklärt ist. Zudem werden Therapien zum Aufhalten der Retina-Degeneration ebenso benötigt wie solche gegen die Neurodegeneration im Gehirn.
Einzig für die spätinfantile NCL-Form CLN2 gibt es seit Juli 2017 eine zugelassene intraventrikuläre Enzymersatztherapie mit dem Wirkstoff Cerliponase Alfa, der eine rekombinante Form des Enzyms TPP1 darstellt, welches bei der CLN2-Erkrankung fehlt.
Da es eben keine präventive oder kurative Behandlungsoption für die meisten NCL-Erkrankungen gibt, liegt der Schwerpunkt auf der palliativen Behandlung klinischer Symptome wie Epilepsie, Myoklonien und Spastiken.
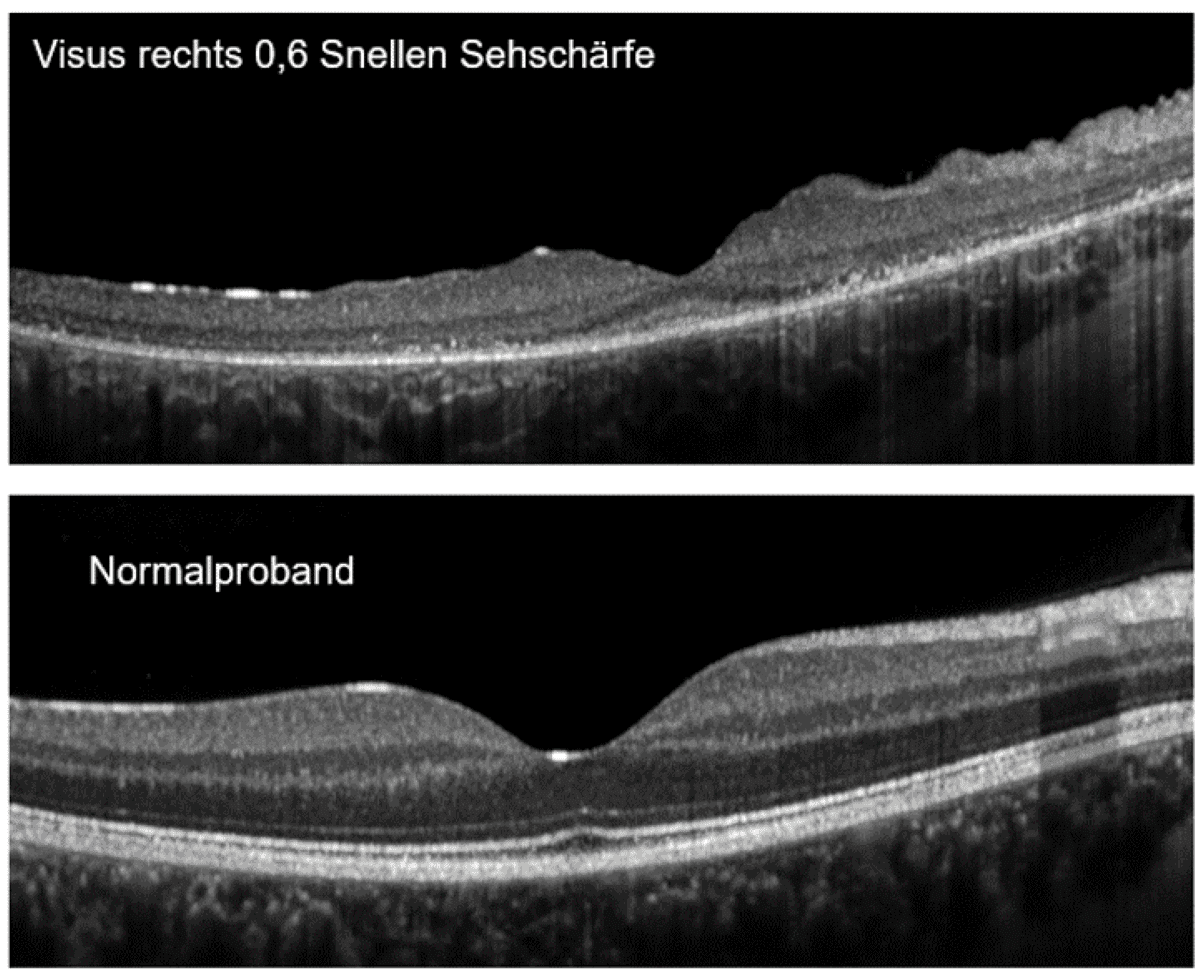
Spektrale Optische Kohärenztomographie (OCT) bei 10-jähriger Patientin mit juveniler NCL: Alle Netzhautschichten sind deutlich verändert.
© Prof. emerita Dr. med. Birgit Lorenz, Senior Clinical Expert Universitäts-Augenklinik Bonn
Warum ist es trotzdem wichtig, dass die Erkrankung so früh wie möglich erkannt wird?
Zunächst ist es für die betroffenen Familien wichtig, so schnell wie möglich eine definitive Diagnose zu erhalten, damit ihnen eine leidvolle Ärzte-Odyssee und eine unnötige Diagnostik erspart bleiben, was bei seltenen Erkrankungen häufig noch die Regel ist. Auch ist so die Möglichkeit für eine frühzeitige humangenetische Beratung gegeben, ebenso wie ein möglichst früher Beginn einer klinischen Behandlung. Familien können sich darüber hinaus von Beginn an über Betreuungs- und Unterstützungsangebote informieren, die den Alltag erleichtern.
Sie sind seit 21 Jahren für die NCL-Stiftung tätig, zunächst als Forschungsleiter, seit 11 Jahren als deren Vorstand. Wie engagiert sich die Stiftung für die Betroffenen und ihre Familien?
Gegründet wurde die NCL-Stiftung im Jahr 2002 von Dr. Frank Husemann, bei dessen Sohn Tim 2001 im Alter von 6 Jahren NCL diagnostiziert wurde. Als Vater eines an NCL erkrankten Sohnes konnte und wollte er nicht tatenlos zusehen, wie ihm durch diese Krankheit Tag für Tag ein wenig mehr von seinem Kind genommen wird. Er wollte sich engagieren und sich hierbei für die nationale und internationale Forschungsförderung einsetzen, um von NCL betroffenen Kindern eine Aussicht auf bisher fehlende Therapie- und Heilungsansätze zu geben. Hierfür initiieren, finanzieren und unterstützen wir als Stiftung Forschungsprojekte und es wird die Bildung eines weltweiten NCL-Netzwerks vorangebracht. Die Ergebnisse sind vielfach auch für neurodegenerative Erkrankungen des Erwachsenenalters von Relevanz. Zudem leisten wir Aufklärungsarbeit bei Ärzten und sensibilisieren die Öffentlichkeit für die Erkrankung.
Die Forschung an seltenen Krankheiten hat oftmals den Ruf des Nischendaseins. Wie Sie gerade erwähnt haben, ist es aber so, dass diese Arbeit auch einen Beitrag leistet zur Grundlagenforschung von anderen Krankheiten.
Ja, die Forschung an seltenen Erkrankungen ist oftmals Pionierarbeit, bei der neuartige Therapien wie die Gentherapie oder eine Enzymersatztherapie das erste Mal getestet werden und zur Anwendung kommen. Daher kann man hiervon sehr viel lernen. Ein vertieftes Verständnis der zellspezifischen Krankheitsmechanismen bei der Kinderdemenz kann zudem dazu beitragen, neue Zielstrukturen für eine Therapie zu identifizieren, die auch bei anderen, häufig vorkommenden neurodegenerativen Erkrankungen wie Alzheimer-Demenz, Parkinson oder Frontotemporaler Demenz relevant sind. Auch könnte die juvenile NCL CLN3 mit dem rasch fortschreitenden Visusverlust als Modell für die altersbedingte Makuladegeneration (AMD) dienen, die der häufigste Grund ist für die Alters-Erblindung in den westlichen Ländern.
Interview: Achim Drucks