Erstaunliche neue Einblicke in die Pathologie des Usher-Syndroms
Seit rund 25 Jahren forscht Prof. Uwe Wolfrum vom Institut von der Johannes Gutenberg-Universität Mainz zum Usher-Syndroms. In Kooperation mit Prof. Reinhard Lührmann vom Göttinger Max-Planck-Institut für biophysikalische Chemie entdeckte er einen neuen Pathomechanismus, der zu dieser Erkrankung führt. Die Wissenschaftler fanden heraus, dass das Usher-Syndrom-1G-Protein SANS eine maßgebliche Rolle bei der Regulation im Splicing-Prozess spielt.
Das humane Usher-Syndrom ist die häufigste Form erblich bedingter Taub-Blindheit. Betroffene können bereits von Geburt an taub sein, können an Gleichgewichtstörungen leiden und mit dem weiteren Fortschreiten der Krankheit ihr Augenlicht verlieren. Die Arbeitsgruppe von Prof. Dr. Uwe Wolfrum vom Institut für Molekulare Physiologie der Johannes Gutenberg-Universität Mainz (JGU) arbeitet seit rund 25 Jahren an der Erforschung des Usher-Syndroms. Sein Team hat nun in Kooperation mit der Arbeitsgruppe von Prof. Dr. Reinhard Lührmann am Max-Planck-Institut für biophysikalische Chemie in Göttingen einen neuen Pathomechanismus entdeckt, der zum Usher-Syndrom führt. Sie fanden heraus, dass das Usher-Syndrom-1G-Protein SANS eine maßgebliche Rolle bei der Regulation im Splicing-Prozess spielt. Des Weiteren konnten sie zeigen, dass Defekte im SANS-Protein zu Fehlern im Spleißen von Genen führen, die mit dem Usher-Syndrom in Verbindung stehen, und dass dadurch möglicherweise die Erkrankung ausgelöst wird.
Rolle von SANS bei der Erblindung bislang noch wenig bekannt
"Wir versuchen, die molekularen Hintergründe aufzuklären, die zur Degeneration der lichtempfindlichen Photorezeptorzellen im Auge bei der Erkrankung am Usher-Syndrom führen", erklärt Wolfrum. Bei einer Erkrankung kann der Gehörverlust mit Cochlea-Implantaten kompensiert werden. Doch gegen die Erblindung gibt es bislang keine Therapien. Für die aktuelle Untersuchung spielt eines der Usher-Syndrom-Proteine, nämlich das USH1G-Protein mit der Bezeichnung SANS, die zentrale Rolle. SANS ist aus vorangegangenen Studien der Arbeitsgruppe als Gerüstprotein bekannt. SANS verfügt dafür über mehrere Domänen, an die andere Proteine andocken können, was die korrekte Zellfunktion gewährleistet. Mutationen im USH1G/SANS-Gen führen zu Fehlfunktionen der auditiven und vestibulären Haarzellen im Innenohr und der Photorezeptorzellen der Netzhaut, die für die sensorischen Defizite von Usher-Syndrom-Patienten verantwortlich sind.
Über die Rolle von SANS im Auge ist wenig bekannt. Kodiert von dem USH1G-Gen wird das Protein in den Photorezeptoren der Netzhaut und Gliazellen gebildet. "Bisher haben wir gedacht, dass SANS als Gerüstmolekül an Transportprozessen im Zellplasma beteiligt ist", so Wolfrum. "Aber jetzt hat mein Mitarbeiter Adem Yildirim im Rahmen seiner Doktorarbeit im Internationalen PhD-Programm Mainz entdeckt, dass SANS mit Splicing-Faktoren interagiert und dadurch das Spleißen von prä-RNA reguliert."
 |
 |
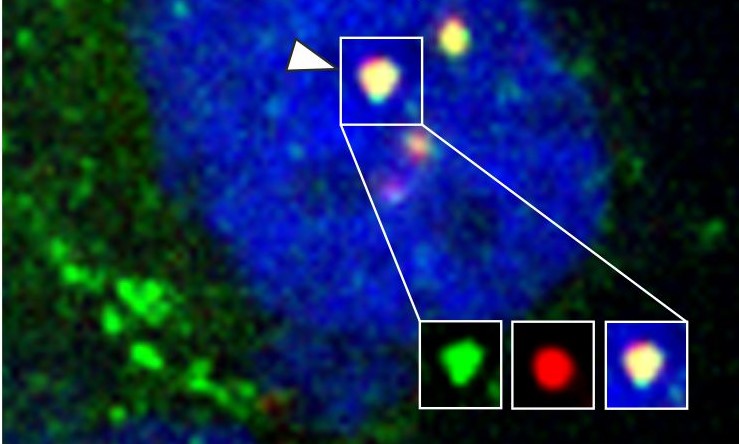
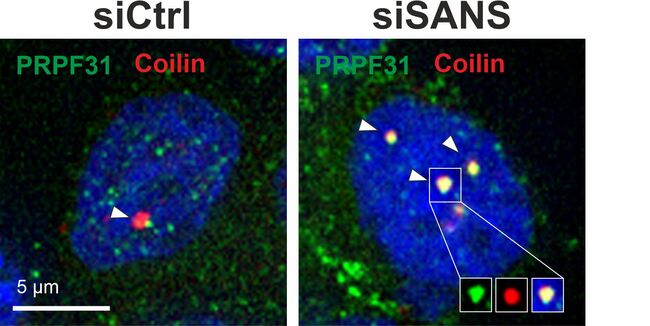
| Bei Abwesenheit oder Herunterregulierung von SANS (siSANS) werden im Gegensatz zur Kontrolle (siCTRL) tri-snRNA-Komplexe nicht aus den Cajal-Körpern (Coilin, rot) entlassen und akkumulieren dort (tri-snRNA-Komponente PRPF31, grün). Foto/©: Uwe Wolfrum/Adem Yildirim | SANS ist für die Freisetzung von tri-snRNPs aus den Cajal-Körpern, deren Transfer zu den Nuclear Speckles, den dortigen Zusammenbau des Spleißosoms und dessen Aktivierung notwendig und damit für das korrekte prä-RNA-Spleißen insgesamt essentiell. Foto/©: Uwe Wolfrum |
SANS reguliert das Spleißen von prä-mRNA
Spleißen, auch als Splicing bezeichnet, ist ein wichtiger Vorgang auf dem Weg vom Gen zur Biosynthese des kodierten Proteins. Dabei werden aus der zunächst transkribierten prä-mRNA die nicht-kodierenden Introns beziehungsweise beim alternativen Spleißen auch für die spätere Proteinvariante nicht benötigte Exons entfernt. Die daraus resultierende mRNA wird anschließend für die Proteinbiosynthese verwendet. Der Splicing-Prozess wird im Zellkern durch das Spleißosom katalysiert, einer dynamischen molekularen Maschine hoher Komplexität, die sukzessive aus zahlreichen Subkomplexen aufgebaut wird, bestehend aus Protein- und RNA-Komponenten.
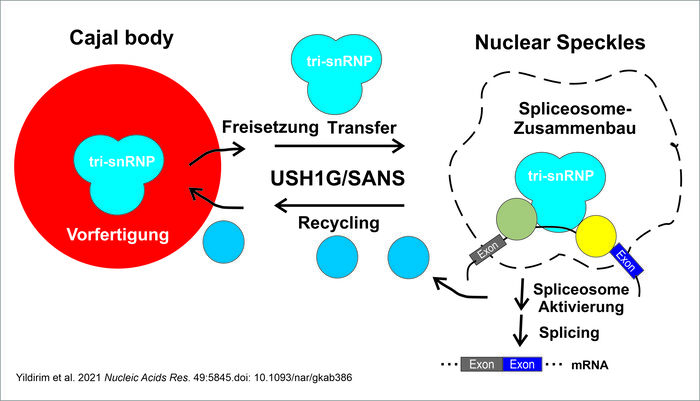
"Nun haben wir überraschenderweise gefunden, dass SANS auch im Zellkern aktiv ist und dort den Splicingprozess modulieren kann", beschreibt Wolfrum die Forschungsergebnisse, die in der Wissenschaftszeitschrift Nucleic Acids Research veröffentlicht wurden. Im Zellkern ist SANS für den Transfer des tri-snRNP-Komplexes, einem Subkomplex des Spleißosoms, aus den Cajal-Körpern, der Vorfertigungsfabrik des Komplexes, zu den sogenannten "Nuclear Speckles" verantwortlich. In diesem Kompartiment binden tri-snRNP-Komplexe an den Spleißosom-Apparat, um diesen nachfolgend zu aktivieren. Zudem dürfte SANS am Recycling der tri-snRNP-Komponenten zurück zu den Cajal-Körpern beteiligt sein.
Die Abwesenheit von SANS und pathogene Mutationen des USH1G/SANS-Gens verhindern den korrekten Zusammenbau des Spleißosoms und dessen sequenzielle Aktivierung. Dies unterbindet das korrekte Spleißen von anderen Usher-Syndrom-relevanten Genen und dürfte schlussendlich zu deren Dysfunktion und somit zur Erkrankung führen. "Wir liefern den ersten Hinweis, dass eine Dysregulation des Spleißens an der Pathophysiologie des Usher-Syndroms beteiligt ist", fassen die Autoren ihre Forschungsergebnisse zusammen. Prof. Dr. Uwe Wolfrum führt weiter aus: "Neben den neuen Erkenntnissen zum Splicing-Mechanismus haben wir auch neue Ziele identifiziert, die wir für eine zukünftige Behandlung und Therapie des Usher-Syndroms nutzen möchten."